










Что такое фенилкетонурия (ФКУ)
Методические рекомендации по диагностике и лечению ФКУ любезно предоставлены специалистами Краковского детского госпиталя
Фенилкетонурия (ФКУ) – это врожденное генетически обусловленное нарушение метаболизма незаменимой аминокислоты фенилаланина (ФА), которая поступает в организм с пищевым белком, в тирозин (ТР). В нашей стране частота этого заболевания невелика: один больной ребенок приходится на пять-шесть тысяч здоровых новорожденных. Причиной заболевания является мутация в гене гидроксилазы фенилаланина (ген ФАГ). В организме больного ребенка происходит накопление избыточного количества фенилаланина в периферической крови, а следовательно, и в ценральной нервной системе (ЦНС). Избыток фенилаланина оказывает тяжелое токсическое действие на организм ребенка. При отсутствии своевременной ранней диагностики это неотвратимо ведет к тяжелому умственному и физическому недоразвитию, являясь причиной ранней детской инвалидности.
Согласно данным Министерства здравоохранения Украины за 2000 год, содержание одного больного с фенилкетонурией, начиная от периода новорожденности до 15 года жизни при полном диетическом лечении, достигает суммы 150 000 долларов США. Расходы государсва на содержание ребенка-инвалида без учёта расходов на скриниговые программы и диетическое лечение составляют около 11 000 гривен. Благодаря эффективному скринингу, а также своевременному внедрению лечения в большинсте случаев становится возможным предупреждение появления неврологических и психиатрических нарушений, что в результате способствует нормальному физическому и психическому развитию больных с ФКУ, а в будущем рождению у них здорового потомства.
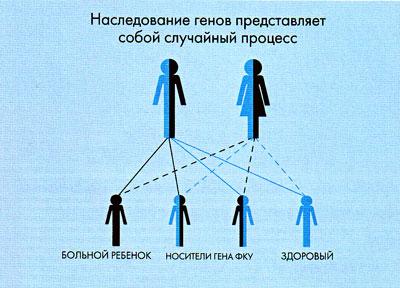 Больной ребенок с фенилкетонурией может родиться только в той семье, где оба родителя являются носителями гена ФКУ. Определить носительство гена ФКУ у родительской пары возможно при генетическом обследовании на гетерозиготное носительство, проводимое в медико-генетических центрах. Если в семье уже есть больной ребенок с ФКУ, носительство гена ФКУ у родителей очевидно. Однако даже если оба родителя являются носителями гена ФКУ, их дети не обязательно будут больны. Если принять за 100% всех детей, которые гипотетически могут родиться в данной семье, можно говорить о следующем риске возникновения заболевания ФКУ:
Больной ребенок с фенилкетонурией может родиться только в той семье, где оба родителя являются носителями гена ФКУ. Определить носительство гена ФКУ у родительской пары возможно при генетическом обследовании на гетерозиготное носительство, проводимое в медико-генетических центрах. Если в семье уже есть больной ребенок с ФКУ, носительство гена ФКУ у родителей очевидно. Однако даже если оба родителя являются носителями гена ФКУ, их дети не обязательно будут больны. Если принять за 100% всех детей, которые гипотетически могут родиться в данной семье, можно говорить о следующем риске возникновения заболевания ФКУ:
- риск рождения больных детей с ФКУ составляет 25%;
- риск рождения детей, являющихся, подобно их родителям, носителями гена ФКУ составляет 50%;
- в остальных 25% случаев родятся здоровые дети
Известно, что частота носительства мутантного гена ФКУ среди населения Украины составляет 2 —3%. Частота заболевания примерно 1:7000.
Ребенок с ФКУ рождается без каких-либо проявлений заболевания. Однако с началом кормления, при поступлении в организм белка грудного молока или его заменителей возникают первые симптомы, трудно распознаваемые не только родителями, но и педиатрами.
Так, в периоде новорожденности до начала лечения у ребенка с ФКУ возможны такие признаки заболевания: необоснованная вялость или беспокойство; обращают на себя внимание рассеянный, блуждающий взгляд, отсутствие улыбки, слабое двигательное оживление. К 6 месяцам у него выявляется задержка психомоторного развития: он перестает активно реагировать на происходящее; утрачивает способность узнавать мать; не переворачивается на живот; не пытается сесть.
Во втором полугодии жизни родители уже не могут не заметить непонимание речи взрослого, неумение выражать голосом и мимикой ребенка свои переживания. У детей старше трех лет нарастают умственная отсталость, возбудимость, повышенная утомляемость; нарушается поведение, что проявляется в расторможенности, психотических расстройствах.
Часто у нелеченых больных фенилкетонурией моча имеет своеобразный «мышиный» запах. Иногда возникают судорожные приступы различной степени выраженности; экзематозные изменения на коже.
Диагностика фенилкетонурии
 Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия - самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии. К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Министерства охраны здоровья организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории Украины.
Из многочисленных наследственных заболеваний обмена веществ (а их насчитывается не менее 700) фенилкетонурия - самое «благоприятное», поскольку при ранней диагностике возможна полная реабилитация больного и его полноценная адаптация к социальной жизни, чего нельзя достигнуть при многих других видах наследственной патологии. К сегодняшнему дню вопрос ранней диагностики у новорожденных заболевания ФКУ решен. В течение последних лет отшлифовывалась методика массового обследования (скрининга) всех новорожденных в нашей стране по выявлению заболевания фенилкетонурией. Приказом Министерства охраны здоровья организовано обеспечение массового скрининга новорожденных на фенилкетонурию на всей территории Украины.
Скрининг-тест производится не раньше чем на 3 сутки (72 часа) после рождения ребенка. При обнаружении у ребёнка повышенного уровня фенилаланина (гиперфенилаланемии) скриниговая лаборатория согласовывает тактику в отношении каждого пациента с ведущим врачом-генетиком.
При уровне ФА в скриниге:
- 3-8мг% - родителей или опекунов информируют о необходимости выполнения контрольного исследования (колориметрическим методом). Если в котрольном исследовании уровень ФА выше 2мг%, то необходимо оповестить родителей/опекунов и пригласить в письменной форме в специализированный диагностический лечебный центр;
- выше 8мг% - необходимо оповестить родителей/опекунов о результате скринигового теста и пригласить в письменной форме в специализированный диагностический лечебный центр.
Во время первого визита врач-специалист (чаще всего педиатр или генетик) обязан предоставить родителям/опекунам исчерпывающую информацию относительно предполагаемого заболевания, его причин и возможностей лечения.
В связи с существованием ряда причин появления гиперфенилаланемии (фенилкетонурия, нетипичные формы фенилкетонурии и тирозенемия), необходимо провести дифференциальную диагностику. Если подтвердится дефицит гидроксилазы фенилаланина (фенилкетонурия), применяется диетотерапия. В случае, если причиной окажется нарушение обмена веществ иного рода, будут избраны иные методы лечения.
Типы фенилкетонурии
Классическая фенилкетонурия –phe > 1200 μмол/л (20 мг%)
Умеренная фенилкетонурия – phe – 900 -1200 μмол/л (15 - 20 мг%)
Мягкая фенилкетонурия – phe –600–900 μмол/л (10-15мг%)
Мягкая гиперфенилаланинемия (gray zone) - phe – 360 – 600 μмол/л (6 - 10мг%)
Мягкая гиперфенилаланинемия- не нуждающаяся в лечении – phe – 120 – 360 μмол/л (2 - 6мг%)
(NIH PKU Conference report: State of the science and future research needs. Feb.22-23.2012)
Злокачественная гиперфенилаланинемия – недостаточность тетpагидpобиоптеpина (BH4)
Дефицит гидроксилазы фенилаланина (PAH)
- фенилаланин ≤ 7 мг% (6 мг%) не нуждается в лечении – наблюдение!
- фенилаланин > 7 мг% (6 мг%) – низкофенилаланиновая диета
Дефицит BH4 (злокачесивенная ФКУ) - фармакологическое лечение.
Клинические появления злокачественной фенилкетонурии:
- тяжелые и быстро прогрессирующие неврологические нарушения (несмотрия на нормальный уровень ФА в крови вследстве диетотерапии)
- гипотония
- спастический синдром
- атаксия
- нарушение глотания
- беспокойство
- прогрессирующая умственная отсталость
- тяжело купирующиеся приступы судорог – наиболее характерные (до 3 мес. жизни признаки заболевания могут отсутствовать)
- уровни фенилаланина нехарактерные ( высокие – как в классической ФКУ или низкие - как в мягкой гиперфенилаланинемии).
Прогрессирующее повреждение нервной системы при злокачественной фенилкетонурии может вести к смерти ребенка, если заболевание не будет обнаружено и не будет применено соответствующее лечение.
Также о дифференцированной диагностике читайте в статье Рекомендации по лечению фенилкетонурии в Польше
Лечение фенилкетонурии
Лечение в фенилкетонурии состоит в ограничении поступления фенилаланина с естественными продуктами в питании ребенка. Это позволяет предупредить наступление тяжелых нарушений ЦНС и обеспечить нормальное интеллектуальное развитие. Показанием к началу диеты с низким содержанием фенилаланина является определение концентрации ФА в сыворотке крови >7-8мг% (согласно рекомендациям польских и немецких центров). Некоторые центры (английские и Европейской Федерации) рекомендуют начать лечение при концентрации фенилаланина >6,6мг%.
Единственным эффективным методом лечения больных ФКУ является специализированная диетотерапия с момента установления диагноза.
Диета при фенилкетонурии - это:
- Уменьшение дозы фенилаланина согласно индивидуальной толерантности к фенилаланину, что означает уменьшение дозы натурального белка в суточном рационе
- Обеспечение соответствующей для нормального развития дозы белка (дополнительный белок без фенилаланина) из продуктов лечебного питания ФКУ
- Обеспечение соответствующей дозы энергии с использованием специальных низкобелковых продуктов
- Обеспечение соответствующей дозы витамин, макро- и микроэлементов – главным образом из препаратов ФКУ и других источников.
Аминокислотные смеси
 У пациентов с ФКУ количество потребляемого белка из натуральных продуктов не может превысить установленной нормы. В связи с этим у маленьких детей и у старших преобладающая часть потребности в белке, т.е. около 80%, должно быть погашено смесями, не содержащими фенилаланин, обогащёнными минеральными ингредиентами.
У пациентов с ФКУ количество потребляемого белка из натуральных продуктов не может превысить установленной нормы. В связи с этим у маленьких детей и у старших преобладающая часть потребности в белке, т.е. около 80%, должно быть погашено смесями, не содержащими фенилаланин, обогащёнными минеральными ингредиентами.
Суточное потребление фенилаланина из пищевых продуктов должно быть ограничено до такого количества, чтобы контролируемый уровень концентрации фенилаланина в сыворотке крови не превышал „безопасного для ЦНС” уровня, т.е. 2-4мг/дл, это и есть индивидуальная суточная толерантность фенилаланина. С целью полного удовлетворения потребностей ребёнка с фенилкетонурией, и поддержания на допустимом уровне употребляемого из продуктов натурального белка и фенилаланина следует все пищевые продукты отмерять и взвешивать, а также выбирать продукты с самым низким содержанием фенилаланина.
Лечебные аминокислотные смеси и препараты для больных ФКУ
Подробнее о методике расчета и коррекции диеты читайте в разделе Диета.
Специальная диета с низким содержанием фенилаланина обычно предотвращает неблагоприятные эффекты, сопровождающие это заболевание. Чем дольше ребенок находится на диете, тем лучше его интеллектуальное развитие. При неадекватном расширении или отмене диеты в подростковом возрасте негативное действие фенилаланина на организм не прекращается. Интоксикация может привести к снижению обучаемости, поведенческим проблемам, вплоть до возникновения психических расстройств. Поэтому в некоторых городах Украины, России и зарубежом принято соблюдение диеты до 18-летнего возраста. Последние европейские и американские наблюдения говорят о том, что в случае классической и некоторых других формы ФКУ желательно пожизненное соблюдение диеты.
Особенно важно знать, что девушки, вступающие в детородный возраст, должны придерживаться диеты до и во время беременности. При несоблюдении женщиной с ФКУ специфической диеты во время беременности у ребёнка возникают следующие пороки развития:
- в 92% случаев - умственная отсталость
- в 73% случаев - микроцефалия
- в 12% случаев - врождённые пороки сердца
- в 40% случаев - низкая масса тела при рождении
Если женщина с фенилкетонурией как минимум за 2 месяца до беременности и во время беременности придерживается диеты и уровень ФА не ревышает 4 мг%, ребенок родится здоровым.
Читайте так же: Как жить с фенилкетонурией